Source: Wen et al. (2008). Following translation by single ribosomes one codon at a time. Nature 452:598-603.
In terms of methodology, this is a very interesting paper. The results, on the other hand, can revolutionize our way of thinking regarding the ribosomes and the translation process. The authors employ the laser trap technology, formerly developed to measure the molecular forces (piconewtons) and displacements (nanometers) generated by a single myosin molecular motor as it interacts with a single actin filament, to follow a single ribosome translating an mRNA hairpin. Below, you can see their experimental set-up.
 According to their results, translation does not occur continuously as we thought so. When the ribosome starts translocating, it opens up the hairpin, thus increasing the distance between the two beads which is measurable by the laser trap. As you see in the figure below, the ribosome goes through a translocation-pause cycles (shown with arrows in the top panel).
According to their results, translation does not occur continuously as we thought so. When the ribosome starts translocating, it opens up the hairpin, thus increasing the distance between the two beads which is measurable by the laser trap. As you see in the figure below, the ribosome goes through a translocation-pause cycles (shown with arrows in the top panel).
 This is a really important change in how we look at translation. For example, each of these pause times (that differ in length quite drastically) can act as foci of post-transcriptional regulation.
This is a really important change in how we look at translation. For example, each of these pause times (that differ in length quite drastically) can act as foci of post-transcriptional regulation.
Source: Stayrook et al. (2008). Crystal structure of the λ repressor and a model for pairwise cooperative operator binding. Nature 452:1022-1025.
My first reaction after seeing paper was, "THEY'RE STILL WORKING ON λ". Well, apparently they do and they have some paradigm shifting results. λ (cI) is the key trasncription factor for lysogeny in this bacteriophage. Simultanuous repression and activation of two distinct (and adjacent) promoters by 'alternate pairwise' binding of this protein is the key to the bistability of the lytic-lysogeny dicotomy. For background information, I refer you to wikipedia. The key question that the authors need to answer is the 'alternate pairwise' binding, where a pair of cI dimers can either take on OR1-OR2 or OR2-OR3 (if OR1 is mutated). Our structural knowledge of cI doesn't tell us why there is no OR1-OR2-OR3 trimer. This exactly what the authors explain through solving the structure of λ repressor in its complete dimer structure. Apparently, the inherent symmetry that we have in mind does not exist (see below; left: schematic cI structure, right: what it really looks like).
 This assymetry impedes a third cI dimer from both binding the other cIs and DNA at the same time.
This assymetry impedes a third cI dimer from both binding the other cIs and DNA at the same time.
Source: Orlando et al. (2008). Global control of cell-cycle transcription by coupled CDK and network oscillators. Nature 453:944-947.
This short paper highlights the fact that the periodicity observed in the expression of many genes across the cell cycle, although initially attributed to the CDKs, is in fact additionally controlled by a transcription network. Their basic observation is that in a cyclin-mutant cell, the periodic expression of the genes, although aberrant, is largely intact; whereas, the cells are arrested in the G1-S transition.
 Their results suggest a transcription network in which the transcription factors are sequentially activated in a cyclic manner as an independent clock based on which the cell determines its progression in the cell cycle. This apparent redundancy (i.e. the presence of two oscillators) in this system ensures the robustness of the cell cycle regulation in our cells.
Their results suggest a transcription network in which the transcription factors are sequentially activated in a cyclic manner as an independent clock based on which the cell determines its progression in the cell cycle. This apparent redundancy (i.e. the presence of two oscillators) in this system ensures the robustness of the cell cycle regulation in our cells.
Source: Spalding et al. (2008). Dynamics of fat cell turnover in humans. Nature 453:783-787.
I loved this paper... I thought it was brilliant... First, a couple of known facts:
- Obesity is a major issue. We might experience a decrease in life expectancy solely based on this factor.
- Fat mass is a function of both the volume and the number of adipocytes.
- The number of adipocytes, although different in individuals, stays the same throughout adulthood.
- The number of adipocytes is set during childhood and adolescence.
- While the number is constant, the turnover is not known.
This paper has tackled the last issue... The fact that the number of adipocytes doesn't change is not equivalent to the concept that these cells are immortal. We do know that these cells go through apoptosis and sometimes even necrosis in vitro but there are no data regarding this matter in living humans mainly due to the toxicity of the methods used for following cell progenies. In this paper, authors have used a modern and intersting method to estimate the rate of turnover in adipocytes. This method is based on the 14C level in the atmosphere. Following the nuclear bomb tests of the Cold War era, a rapid increase and notable increase in 14C level of the atmophere was reported. Upon the declaration of Test-Ban Treaty (1963), this level has decreased exponentially because 14C is assimilated by the plants as biomass which is then transferred to other ecological levels (including us). For example, someone who was 20 years old when those tests happened should have a pre-Cold War level of 14C if the adipocytes are immortal and don't regenerate. However, the measurements in this study prove otherwise. Integrating data from many samples followed by a mathematical modeling, the authors estimate an annual turnover rate of 10% for fat tissue.
The fact that 10% of the adipocytes are renewed annually is very important for developing drugs against obesity. If we can tackle the birth rate or increase the death rate then (theoretically) we should be able to control total body fat.
Source: Guelen et al. (2008). Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453:948-951.
This is a great paper with interesting methods and exciting results. The authors use DamID to identify the regions of the genome that interact with the nuclear lamina (NL). For this they make a DNA adenine methyltransferase (Dam) fused to human lamin B1. Upon formation of the nuclear structure, Dam methylates the nearby adenine nucleotides that can be later identified as foci of interaction. Upon mapping genome-NL interactions (lamina associated domains or LADs) they compare the NL-free regions with those that are attached. Their first observation is that NL-bound regions have a low frequency of coding sequences, and those that are there are generally expressed at low levels. Secondly, NL-bound regions are isolated from the other parts of the genome through insulator proteins, promoters directed away from the LADs and GC boxes.
This paper makes the case for a complex level of chromosome organization where domains can be seperated by LADs and thus acting as perfect insulators.
Source: McMurray et al. (2008). Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature 453: 1112-1116.
First of all, a couple of my friends have started a similar blog on parasitology (http://parasitediary.wordpress.com/); for those of you who are working in this field, I think they'll be thrilled if you visit their blog, leave comments or even better contribute.
Now back to our own post... well, tumorigenesis is a hot topic with many papers being published in very good journals solely based on a subject profiling. This paper, however, very well deserves its publication in Nature. The authors start with the notion that genes with synergistic effects across tumorigenic mutations should play a key role in the emergence of the transformed state and throughout the paper they provide evidence in support of their hypothesis. First, they profile the gene expressions in p53, ras, and p53/ras cell lines. Then, they focus on the genes in the double mutant with a fold-change larger than the sum of fold changes in the single mutants (i.e. synergistic genes). Note that these fold-changes can be both positive and negative; thus, we include both positive and negative synergism. An initial look at these genes shows a diversity of roles in signaling, transcription, apoptosis and adhesion that are generally deregulated in the available cancer gene-expression datasets.
To show that these genes are important in tumorigenesis, they restore the normal expression levels of 24 synergistic and 14 non-synergistic genes, one by one, in the double mutant background. For this, they employ retro-virus mediated re-expression or shRNA-dependent knockdown. They show that in 14/24 cases, for the synergistic genes, there is a significant reduction in tumorigenesis upon restoring normal expression levels; whereas, only 1/14 non-synergistic genes shows such level of effect.
To summarize, this paper makes the case for synergistic genes to be foci of transformation; thus, excellent genes for studying or therapeutic targeting.
Source: Berkman et al. (2008). Evolution and Creationism in America's Classromms: A National Portrait. PLoS Biology 6(5):e124.
"Evolution--more precisely opposition to it--is profoundly important to fundamentalist Christianity, where it has played a critical role in its early formation as doctrine and as a social movement". This is one of the key sentences in this paper, highlighted by the authors themselves. They make the case that despite recent judiciary victories of evolution over its petty imaginative counterpart (Intelligent Design [at least someone is intelligent]) in the courtroom, we (the scientists) are losing the case in the classrooms where the science teachers have to actually teach evolution. While only 16% of the science teachers believe in creationism (compared to 48% of the general public), they have a hard time devoting enough resources to this theory which is dubbed "a unifying theme in science" by the National Science Education Standards. As the authors put it, "community pressures place significant stress on teachers as they try to teach evolution, stresses that can lead them to de-emphasize, downplay, or ignore the topic".
The question is, why do we care? For example Richard Dawkins believes that any public discussion of this matter with creationists would bring them a sort of validity that they don't have and don't deservce (thus the question mark in the title of this post). Everyone treat this matter as if it is a personal decision to make. Believeing the Earth to be flat is not a personal decision, so is not evolution. Nowadays, science is pretty much speclizied with researchers deep into their specific knowledge. For example, I trust my friends in Math department when they say "the world has 11 dimensions" because I don't know squad about string theory. Or I don't question my doctor's diagnosis as absurd as they may seem to my untrained eyes (e.g. you go to them for headaches, they say there's something wrong with your stomach).... The same should hold for evolution... We know what we're talking about because we have studied this for years... We have a whole field that's working based on it.... When we say it holds, TRUST ME, it holds.
Source: Dantas et al. (2008). Bacteria Subsisting on Antibiotics. Science 320:100-103.
We know antibiotics as weapons against bacteria...however, not only there are strains that are resistant to these small molecules but also there are ones capable of growing on them as the sole carbon source (which sort of defeats the purpose). However, I should note that I'm not really surprised. There are no dead-end biosynthetic pathways in nature. If a compound is synthesized by an organism, there is always another one who is capable of degrading it. However, antibiotics are really important cases, because we exclusively use them for fighting bacterial infections and through the years the clinical isolates are growing resistant.
This study in the Church lab makes the case that there is a huge reservoir of antibiotic-resistance pathways already present in nature that we should be aware of and be able to deal with in very near future (I sound like an alarmist). As you see in the figure below from the original paper, each of the soil samples can grow on at least one antibiotic as the sole carbon source. Phylogenetic distribution of these bacteria is quite fascinating. The range is diverse and encompasses many known infection agents. These results show the existence of a notable reservoir of genes conferring antibiotic resistance and worse case scenario, treating an infection may boost the chance of another one.
Phylogenetic distribution of these bacteria is quite fascinating. The range is diverse and encompasses many known infection agents. These results show the existence of a notable reservoir of genes conferring antibiotic resistance and worse case scenario, treating an infection may boost the chance of another one.
Source: De Silva et al. (2008). Specific DNA-binding by Apicomplexan AP2 transcription factors. Proc Natl Acad Sci, in press (available online).
This is an outstanding study by our neighbors (Llinas lab) in the Lewis-Sigler Institute. As always, I asked Erandi to write a summary of her paper for me to post here which she accepted. Here it comes...
Our lab studies the malaria-causing Apicomplexan parasite Plasmodium falciparum. Bioinformatic analysis of the parasite genome demonstrates a dearth of specific transcription factors that could modulate the periodic gene expression cascade seen during the red blood cell stages of development. Furthermore, computational prediction of cis-regulatory elements has proven difficult given the extensive A-T content of intergenic regions (approaching 90%) in this unusual organism. Up to now, only a handful of regulatory elements sufficient to drive gene expression have been experimentally characterized in P. falciparum, and their cognate DNA-binding proteins remain unknown. Our recent work characterizes the in vitro DNA-binding specificities of two members of a recently identified Apicomplexan AP2 (ApiAP2) family of putative transcriptional regulators from Plasmodium falciparum. The ApiAP2 proteins contain AP2 domains homologous to the well-characterized plant AP2 family of transcriptional regulators, which play key roles in development and environmental stress response pathways. We assayed ApiAP2 protein-DNA interactions using protein binding microarray technology (Berger et al, 2006. Nat Biotechnol 11:1429–1435) and combined these results with computational predictions of co-expressed target genes to couple these putative trans factors to corresponding cis-regulatory motifs in Plasmodium. We also showed that the protein-DNA sequence specificity is conserved in orthologous proteins between phylogenetically distant Apicomplexan species. Remarkably, our experimentally-derived cis-regulatory motifs closely match independent computational predictions for motifs involved in stage-specific gene regulation in P. falciparum (Elemento et al, 2007. Mol Cell 28:337–350). This study represents the first characterization of the DNA-binding specificities of putative transcriptional regulators in Plasmodium and lays the foundation for the exploration of the role of ApiAP2 proteins during parasite development.
 DNA motifs specifically bound by AP2 domains predicted using protein-binding microarrays (PBMs) and computational analysis. (A) The core nucleotides (boxed) in the motif specifically bound by the P. falciparum AP2 domain of PF14_0633 are identical to those bound by its Cryptosporidium parvum ortholog cgd2_3490 (top two rows). The motifs determined from the PBM correspond to motifs predicted by computational analysis of 5’ upstream regions of co-expressed genes (bottom row). (B) The PBM-predicted motif bound by the tandem AP2 domains of PFF0200C (top row) is identical to the motif bound by the first domain alone (middle row). Domain 2 of PFF0200C did not bind a specific DNA motif (data not shown). Both PBM-predicted motifs for PFF0200C match the computationally predicted motif (bottom row).
DNA motifs specifically bound by AP2 domains predicted using protein-binding microarrays (PBMs) and computational analysis. (A) The core nucleotides (boxed) in the motif specifically bound by the P. falciparum AP2 domain of PF14_0633 are identical to those bound by its Cryptosporidium parvum ortholog cgd2_3490 (top two rows). The motifs determined from the PBM correspond to motifs predicted by computational analysis of 5’ upstream regions of co-expressed genes (bottom row). (B) The PBM-predicted motif bound by the tandem AP2 domains of PFF0200C (top row) is identical to the motif bound by the first domain alone (middle row). Domain 2 of PFF0200C did not bind a specific DNA motif (data not shown). Both PBM-predicted motifs for PFF0200C match the computationally predicted motif (bottom row).
Source: Cardinale et al (2008). Termination Factor Rho and Its Cofactors NusA and NusG Silence Foreign DNA in E. coli. Science 320: 935-938.
In E. coli, the termination of transcription is achieved through either the formation of stem-loops in the nascent mRNA or the activity of an RNA-DNA helicase known as Rho. In this paper, the authors reveal the global role of this protein as a regulator of transcription in a genome-wide scale. The antibiotic Bicyclomycin (BCM), a potent inhibitor of Rho, is employed to knock-down the activity of this protein (rho is an essential gene) followed by gene-expression microarrays to assess the mRNA content of the cell.
The most prominent observation, in the absence of Rho, is the significant up-regulation of horizontally transfered genes along with prophages in the genome. 2D protein gels, however, show a limited deregulation in the protein content where the differences are limited to an upregulation of anaerobic genes and a number of acid response genes (e.g. cadA).
Deleting the dormant prophages in the genome (e.g. rac) drastically increases the resistance to BCM, emphasizing the role of Rho in suppressing these invasive foreign DNA fragments.
Apart from its exciting results, I like this paper for another reason. Have you ever read a paper and said, "damn it, I had this idea...". In this case, apart from the ideas, I even had the necessary strains and a batch of BCM and I was ready to do this experiment. However, a discussion with my adviser suggested that this was too much off the path from my own project.... and a year later... here it is... published in SCIENCE :)
Source: Najafabadi and Salavati (2008). Sequence-based prediction of protein-protein interactions by means of codon usage. Genome Biology 9:R87
My dear friend Hamed, now working in the Parasitology department of McGill University, has published a recent paper in Genome Biology which (believe it or not ;)) has made the highly accessed set in this rather prestigious journal. I asked him to write a summary of his paper and despite his tight schedule (believe me, it's TIGHT), he sent me the following. ENJOY.
The mainstay research in our lab is focused on trypanosomatids, human parasites with almost-newly sequenced genomes. Their distant homology with well-studied organisms such as yeast, bacteria and animals makes it extremely difficult to draw any conclusions regarding the functions of the majority of their genes using homology-based methods. Although this paper does not even mention the trypanosmatids, it describes a method that we basically developed for prediction of functional linkages between proteins in these parasites. We surprisingly found that codon usages of the proteins that either physically or functionally interact with each other share more similarities than the codon usages of unrelated proteins. This may be due to factors such as the requirement for similar expression levels of interacting protiens, physical proximity of the genes of the interacting proteins on genome, or unknown evolutionary forces yet to be determined. We developed a classifier that uses codon usage for prediction of physical/functional interactions of proteins. Most notably, we found that combining this classifier with previously published classifiers almost doubles their sensitivity. We also showed that, no matter which organism, once a suitable training set is available this method is the first choice for homology-independent prediction of functional and physical interactions.
Source: Hillenmeyer et al. (2008). The Chemical Genomic Portrait of Yeast: Uncovering a Phenotype for All Genes. Science 320: 362-365.
In the rich media generally used in the labs for growing yeast cells (e.g. YPD), in fact, ~80% of the yeast genome is dispensable with no evident phenotypic effect on growth. While this can be in part attributed to redundancy and robustness in the biological systems, we know that there aren't actually that many redundant pathways (let alone 80% of the genome). The most evident line of reasoning goes through the definition of "essentiality". Yeast cells, in their natural habitats, hardly ever encounter such a perfect environment such as a rich medium. Thus, the repertoire of seemingly non-essential genes, most probably, is essential in natural habitats and in different stress conditions.
In this paper, the authors have tested 1144 perturbtation conditions to identify the genes with phenotypic effects in homozygous and heterozygous contexts. It should be noted that although I'm not a fan of doing these experiments myself, I think they should be done (ofc0urse not by me). In their findings the authors report only 3% of genome with no growth effect in all these conditions. They show that most of these genes are generally inactive. Now, if you're interested in a certain gene, you should go to this database and do your study in a condition which bears a phenotype from this gene.
Apart from the concept of essentiality, they also look into the genes that are technically labeled as "Multi-Drug Resistance" genes. They define a set of 51 genes in this category which includes a set of known MDR genes.
Source: Nagalakshmi et al (2008). The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing. Science 320:1344-49.
Enfin, the long anticipated RNA-seq paper. Throughout the last decade, gene-expression microarrays have been vastly employed for reporting changes in the expression level of all the transcripts. However, higher efficiency and lower prices in the sequencing market may very well render these methods obsolete in a matter of years. cDNA microarray chips don't provide any information regarding the 5' and 3' end of the transcript (important for e.g. alternative splicing). Although, one might argue the usage of tiling arrays for such matters, 97 slides for a human genome is out of question.
In the RNA-seq method, the mRNAs are extracted (using oligo-dT columns), converted to cDNA, subjected to fragmentation and massive high-throughput sequencing (Illumina sequencing which gives ~35 nucleotides from one end). I'll not go through the proof-of-principle steps, but suffices to say that the method works quite decently. 91.5% of the genes were active with 20% of them reported as highly expressed (biosynthetic pathways and ion transporters). In this type of data, a sharp decline in the number of reads denotes that 5' and 3' ends. The authors have calibrated their parameters using 5' RACE followed by sequencing for 125 genes.
Aside from boundary information, this approach enables us to comment on many other issues (e.g. uORFs, polyA sites and ...). As I said, my guess is that this approach will become mainstream in molecular biology in near future.
Source: Sheari et al. (2008). A tale of two symmetrical tails: Structural and functional characteristics of palindromes in proteins. BMC Bioinformatics 9:274.
This is a paper from my dear friend Amir. He has kindly written a summary of his work for me which I have pasted below.
Well, this is a short paper, but it was really difficult to do all those analyses. In this paper, we showed that palindromic sequences in proteins are prevalent because they coincide with low-complexity regions. In addition, we showed that they have a high tendency to form alpha-helices. This is already an important result, because it is generally believed that the existence of low-complexity regions coincides with lack of stable secondary structures (=formation of intrinsically disordered proteins). This paper clearly shows that palindromes in proteins are exceptions to this rule.

Source: Tarassov et al (2008). An in Vivo Map of the Yeast Protein Interactome. Science 320:1465-70.
This paper is interesting in the sense that protein interactions are assayed in vivo and in the context of living cells. The method is quite interesting with room for extensive future tweaking. The authors fuse the proteins to bait and prey fragments of murine dihydrofolate reductase (mDHFR). Upon forming an interaction between the two proteins, mDHFR forms a functional enzyme enabling the cell to grow in the presence of methotrexate (see figure below).
 The authors succeed in fusing F[1,2] to 75% of MATa ORFs and 83% of MATalpha (total coverageof 93%). Then they mate these strains in a pairwise set-up and record the growth in the presence of methotrexate as a proxy for interaction between proteins. I haven't gone through their results carefully but it seems like they do capture most of the known gold standard datasets. This was a fun read, congratulations to the authors for their hard work in getting all these fusions....
The authors succeed in fusing F[1,2] to 75% of MATa ORFs and 83% of MATalpha (total coverageof 93%). Then they mate these strains in a pairwise set-up and record the growth in the presence of methotrexate as a proxy for interaction between proteins. I haven't gone through their results carefully but it seems like they do capture most of the known gold standard datasets. This was a fun read, congratulations to the authors for their hard work in getting all these fusions....
Source: Acar et al. (2008). Stochastic switching as a survival strategy in fluctuating environments. Nature Genetics 40(4):471-475.
Previously, I had a post regarding phenotypic differences in populations with identical genotypes (Dr Jeckyll and Me Hyde). There, we made the case that persistence, as a switching phenotype, can help isogenic populations survive adverse changes in the environment. Here, the authors have modeled this problem in a more general format implying that the stochastically switchable phenotypes counteract the fluctuations in the environment.
The authors have engineered the GAL bistable switch in yeast to design two distinct strains: fast switchers and slow switchers. They have also envisioned two different environments (E1 and E2) each of which are suitable for one of these two switching paces (see figure below).
 Using both mathematical and experimental models, the authors show that fast switching cells hold an advantage in rapidly changing environments while slow switchers do better in more stable conditions. In other words, the pace of stochastic switches can evolve in response to the fluctuations in the environment as an adaptive response for higher survival rate.
Using both mathematical and experimental models, the authors show that fast switching cells hold an advantage in rapidly changing environments while slow switchers do better in more stable conditions. In other words, the pace of stochastic switches can evolve in response to the fluctuations in the environment as an adaptive response for higher survival rate.
Source: Mettetal et al. (2008). The frequency dependence of osmo-adaptation in Saccharomyces cerevisiae. Science 319: 482-484.
This is a very fun paper for someone with my interests in this field. An MIT-Harvard coalition of forces is employed to reverse engineer a well-studied pathway in yeast and to define the dynamics of the cellular system. Below, the MAP kinase pathway is shown which leads to the activation of Hog1 (a transcription factor) upon osmotic stress and in turn its localization to the nucleus. Once inside the nucleus, Hog1 changes the expression level of many genes with roles in stress response.  In this study, the authors employ a microscopy based technique to measure Hog1-YFP nucleus localization in response to osmotic shock (the response in normalized using Nrd1-RFP, a strictly nuclear protein). This set-up is employed in a chemostat with stepwise addition and depletion of medium containing excess NaCl. The figure below (from the original paper) shows this experimental set-up.
In this study, the authors employ a microscopy based technique to measure Hog1-YFP nucleus localization in response to osmotic shock (the response in normalized using Nrd1-RFP, a strictly nuclear protein). This set-up is employed in a chemostat with stepwise addition and depletion of medium containing excess NaCl. The figure below (from the original paper) shows this experimental set-up.
 The authors record the responses in wild-type and pbs2 mutants. Reduced expression of Pbs2 decreases the level of Hog1 and enables them to measure the Hog1 independent feedback loops. They interpret a successful predictive model from the data and based on the fitted parameters they comment on the dynamics of the pathway. First of all, the cell recovers after 5 min which is much lower than the time needed for gene-expression based changes (>15min). Then, why is Hog1 needed to activate and deactivate so many genes? The authors address this by inhibiting protein synthesis using cycloheximide. Their results shows that the hog1 induced changes in protein levels render the cells more competent in recovering from the shock in the future challenges. In other words, Hog1 activation carves a short memory of osmotic shock in the cell.
The authors record the responses in wild-type and pbs2 mutants. Reduced expression of Pbs2 decreases the level of Hog1 and enables them to measure the Hog1 independent feedback loops. They interpret a successful predictive model from the data and based on the fitted parameters they comment on the dynamics of the pathway. First of all, the cell recovers after 5 min which is much lower than the time needed for gene-expression based changes (>15min). Then, why is Hog1 needed to activate and deactivate so many genes? The authors address this by inhibiting protein synthesis using cycloheximide. Their results shows that the hog1 induced changes in protein levels render the cells more competent in recovering from the shock in the future challenges. In other words, Hog1 activation carves a short memory of osmotic shock in the cell.
Source: Silva et al. (2008). Profiling Essential Genes in Human Mammary Cells by Multiplex RNAi Screening. Science 319: 617-620.The method used in this paper is conceptually very similar to a method used in our lab for whole-genome fitness assays. The stated problem is to make single gene knock-out and measure their growth. However, it is not possible to delete the genes one by one in a cell-line. So shRNAs have been the method of choice for this study. They start with an shRNA plasmid library, package the plasmids into a virus and infect the cells to have a library with single genome insertions. In these infected cells, the expression of a gene is knocked down through miRNA like processes.
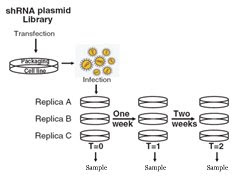 After taking samples at several timepoints of the selection period, they PCR up the constructs, label them and compare them to the unselected library on a microarray chip (see below).
After taking samples at several timepoints of the selection period, they PCR up the constructs, label them and compare them to the unselected library on a microarray chip (see below).
 Using this approach, the authors identified and later validated essential genes in several human cell-lines.
Using this approach, the authors identified and later validated essential genes in several human cell-lines.
Source: Karpova et al (2008). Concurrent Fast and Slow Cycling of a Transcriptional Activator at an Endogenous Promoter. Science 319:466-469.
Personally I am not interested in the kinetics of promoter activation, I just care about the underlying sequence motifs. However, I found the methods used in this paper utterly exciting and very well worthy of our time to read. I'll just go through the basic idea; for more details consult the original paper.
The authors set out to study the kinetics of promoter occupancy in endogenous promoters. To do so, they use the CUP1 promoter in yeast which is found in 10 tandem repeats. Adding three GFPs to Ace1p (the cu-inducible TF which binds CUP1) makes it possible to actually visualize the occupancy under a microscope (see the figure below with and without cu induction).
 Upon making this set-up, they use laser beams to bleach the transcription factors occupying the promoter (yellow arrows above). Then they measure the time that is needed for the fluorescence to reach the initial intensity. This graph (below) depends on the retention time of the activator on the promoter and in this case the intensity reaches maximum in a matter of minutes.
Upon making this set-up, they use laser beams to bleach the transcription factors occupying the promoter (yellow arrows above). Then they measure the time that is needed for the fluorescence to reach the initial intensity. This graph (below) depends on the retention time of the activator on the promoter and in this case the intensity reaches maximum in a matter of minutes. As I said, I won't go into details, but using these tools the authors make the case that this activator goes through a fast (~1 min) and a slow (~15-90 min) cycle. They show that (using ChIP-chip and RT-PCR) the slow cycling regulates the quantity of the RNA produced; whereas, the fast cycle is actually in charge of transcription initiation.
As I said, I won't go into details, but using these tools the authors make the case that this activator goes through a fast (~1 min) and a slow (~15-90 min) cycle. They show that (using ChIP-chip and RT-PCR) the slow cycling regulates the quantity of the RNA produced; whereas, the fast cycle is actually in charge of transcription initiation.
Source: Park et al. (2008). Fine mapping of regulatory loci for mammalian gene expression using radiation hybrids. Nature Genetics 40(4):421-429.
This is a very interesting paper with exciting methods and significant results. I wanted to write about it but instead I decided to ask Joshua Bloom, a good friend of mine and a graduate student in our department, who is the third author of this paper to write a post instead. What follows is a summary of this paper in Josh's words which I truly appreciate.
Until now, the identification of expression QTLs (eQTLs) has relied on segregating polymorphisms in a population. In this paper the authors develop a new approach for understanding the genetics of mammalian gene expression using a panel of mouse-hamster radiation hybrid cells. The introduction thoroughly describes the history and properties of these cells. In brief, each cell has a different random ~30% of mouse DNA split up into 10Mb fragments in the background of a hamster cell line. The advantages of using this panel is that it has an order of magnitude more breakpoints than mouse recombinant inbred strains and is not susceptible to long stretches of linkage disequilibrium, thus allowing finer localization of regulatory loci.
The idea behind the QTL mapping in the context of radiation hybrids is determining what effect the presence or absence of a copy of a mouse gene has on either its own expression (cis) or on the expression of any other gene (trans) in the genome. As expected, considering known effects of gene dosage on expression, the authors find that most genes show significant cis ceQTLs. However, the effect sizes are not proportional to the increase in gene dosage. Also, the effect sizes for cis ceQTLs on the X chromosome are significantly decreased compared to the effect sizes of cis ceQTLs on autosomes. This suggests that expression of X chromosome genes may be less sensitive to gene dosage. Interestingly, the authors identify ~ 30,000 trans ceQTLs, the majority of which induce the regulated gene. They also identify hotspots of regulation and verify one hotspot with a transfection experiment. Although many of the GO categories enriched in trans ceQTLs were identified as transcription related, many other categories were not.
The significant linkages will be a useful community resource for biologists and this approach provides a new strategy for understanding genetic regulation in mammals.
Source: Tagkopoulos et al (2008). Predictive Behavior Within Microbial Genetic Networks. Science 320:1313-17.
Whole-genome fitness assays don't match gene expression microarrays. For example, a heat shock in E. coli results in up- and down-regulation in a range of genes; however, many of these changes are dispensable. Our lab makes the case that the transcription network makes many changes not based on necessity but based on anticipation.
Artificial activation of the envelop stress response in turn activates cytoplasmic heat shock response, not because it contributes to protein folding in periplasm but because higher temperature outside the cell is coupled with higher temperature in cytoplasm. E. coli uses the data from periplasm to anticipate a future change in its cytoplasm. This is a very basic associative learning (or classical conditioning). A graduate student and a postdoc in our lab have elegantly demonstrated the ability of transcription networks to incorporate environmental signals into effective anticipations. This study has both computational and experimental facets. First, Ilias developed a simulation framework called Evolution in Variable Environments (EVE) to show that transcription network models are very well capable of learning simple associations (AND, OR, XOR and etc.). In parallel, Yirchung Liu studied the negative correlation between the oxygen level and tempertaure in the natural habitat of E. coli as it passes through the mammalian GI tract to the outside world and vice versa. She showed that increasing the temperature results in a shift towards anaerobic respiration in wild-type E. coli. Now if you evolve E. coli in an environment in which the correlation between temperature and oxygen level is de-coupled, the organism forgets this correlation and does not switch to anaerobic respiration in higher temperatures.
This case portrays one of the weak points of gene-expression microarray analysis.
Source: Campbell et al (2008). Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nature Genetics 49(6): 722-729.
Without any doubt, this paper excels in portraying the power of massive parallel sequencing to address whole genome problems. The idea is elegant and simple, they nebulize the genomic DNA extracted from two lung cancer cell lines (NCI-H2171 and NCI-H1770 for which the genomic rearrangements are in part identified) while ligating adapters to the sheared ends. The resulting fragments are size selected using gel extraction (200-400bp) and then applied to massive parallel sequencing for reading the two fragment ends. They then aligned the reads back to the genome with the idea that the two end reads falling far from each other (600bp) represent chromosomal re-arrangements. This process is shown here with a figure from the original paper:
 To summarize the results, they succeed in capturing the previously known re-arrangements in these two cell-lines. Using the same data, they also extract information regarding the copy-number and the structure of complex amplicons in the genome. The details do not really matter (see the original paper if you are interested); nevertheless, this paper is a perfect example of combining whole-genome experimental and computational tools to reveal the underlying biological differences.
To summarize the results, they succeed in capturing the previously known re-arrangements in these two cell-lines. Using the same data, they also extract information regarding the copy-number and the structure of complex amplicons in the genome. The details do not really matter (see the original paper if you are interested); nevertheless, this paper is a perfect example of combining whole-genome experimental and computational tools to reveal the underlying biological differences.
Source: Smith EN, Kruglyak L (2008) Gene–environment interaction in yeast gene expression. PLoS Biol 6(4): e83.
This is an elegant and long paper by Kruglyak lab in our institute. Given the magnitude of the results, I'll just breeze through the most interesting parts (from my point of view, of course). The initial goal of the authors is to discover genes whose expression changes in response to environment in a species-specific manner. The figure below (from the original paper) clearly shows this case. As you see, the gene-environment interaction is defined as a strain-condition effect on expression between BY and RM yeast strains. In order to pinpoint the genetic determinants of such strain-specific interactions, the authors have expression profiled 109 segragants from these two parents. Knowing the exact alleles at each locus for all of these segragants enables to identify quantative loci that contribute to each strain-condition effect (using gxeQTL). They can also identify cis acting and trans acting elements based on the linkage to the genes.
As you see, the gene-environment interaction is defined as a strain-condition effect on expression between BY and RM yeast strains. In order to pinpoint the genetic determinants of such strain-specific interactions, the authors have expression profiled 109 segragants from these two parents. Knowing the exact alleles at each locus for all of these segragants enables to identify quantative loci that contribute to each strain-condition effect (using gxeQTL). They can also identify cis acting and trans acting elements based on the linkage to the genes.
As a showcase, the authors characterized IRA2 as a locus responsible for differential expression of many growth-related genes. As they put it themselves, "The RM allele of IRA2 appears to inhibit Ras/PKA signaling more strongly than the BY allele, and has undergone a change in selective pressure". I'll not go through the specifics of this study... you can refer to the original paper for a more detailed analysis.
Source: Pearl S, Gabay C, Kishony R, Oppenheim A, Balaban NQ (2008) Nongenetic individuality in the host–phage interaction. PLoS Biol 6(5): e120.
Individuality is a broad term; however, in genetic context it refers to phenotypic differences between individuals with identical genomes (e.g. identical twins). This paper studies bacteria vs phage interactions in isogenic backgrounds and the non-genetic choices that the entities in the community face individually. A clear example of such behavior is bacterial persistence in adverse environments; certain bacteria survive stresses (e.g. antibiotics) by shutting down the cellular processes and entering a dormant state.
Here, the authors have studied the influence of persistence as a choice in host-phage interactions in populations of bacteria. They have cloned a temperature sensitive cI (lambda repressor) in a lysogenic strain; in this strain, shifting to higher temperatures results in activation of lytic pathway and cell death. They compare the survival rate of wild-type and persister (hipA7 strain) strains upon switching to a higher temperature (which leads to phage activation). They do this through measuring the activity of lytic genes (using GFP reporters cloned downstream of lytic promoters) and tracking cell-fate using live microscopy. The figure below, taken from the original paper, shows the higher survival rate of hipA7 strains. The authors also show that the phages cannot kill persister cells in their dormant state and they rely on the cell to switch back. To test this, they initially lyse all the hipA7 mutants that are active by an incubation in high temperature. Then they measure the exit from dormancy by locating the first occurrence of cell division. They simultaneously measure the activation of lytic pathway by a GFP reporter. Their results show a high correlation between the lytic pathway activity and exit from dormancy.
The authors also show that the phages cannot kill persister cells in their dormant state and they rely on the cell to switch back. To test this, they initially lyse all the hipA7 mutants that are active by an incubation in high temperature. Then they measure the exit from dormancy by locating the first occurrence of cell division. They simultaneously measure the activation of lytic pathway by a GFP reporter. Their results show a high correlation between the lytic pathway activity and exit from dormancy.
Now, what is the significance of these observations? These results clearly show the effect of persistence on host-phage interactions. In other words, a high persister strain (like hipA7) would overcome a low persister (e.g. wild-type) in an environment with frequent appearance of phages; whereas, in normal environments they should do worse given their lower growth rate due to a higher chance of dormancy. The authors actually test this model through competing wild-type and hipA7 strains with different frequency of phage inductions. This figure from the paper perfectly shows the hipA7 overcoming wild-type in high phage induction environments (red is hipA7 and blue is wild-type).

These observations imply that persistence in bacteria acts as a general immune response at the population level to overcome environmental and predator-prey stresses. In my opinion, this random entrance and exit from dormancy in response to predators and phages very much resembles the random strategies evolved in the behavior of higher organisms (from a game-theoric point of view). Simply put, bacterial communities are very well capable of complex behavioral concepts despite the evident lack of the nervous systems.
 According to their results, translation does not occur continuously as we thought so. When the ribosome starts translocating, it opens up the hairpin, thus increasing the distance between the two beads which is measurable by the laser trap. As you see in the figure below, the ribosome goes through a translocation-pause cycles (shown with arrows in the top panel).
According to their results, translation does not occur continuously as we thought so. When the ribosome starts translocating, it opens up the hairpin, thus increasing the distance between the two beads which is measurable by the laser trap. As you see in the figure below, the ribosome goes through a translocation-pause cycles (shown with arrows in the top panel). This is a really important change in how we look at translation. For example, each of these pause times (that differ in length quite drastically) can act as foci of post-transcriptional regulation.
This is a really important change in how we look at translation. For example, each of these pause times (that differ in length quite drastically) can act as foci of post-transcriptional regulation.


















